孟慶男,梁香靈,田娜,湯玉斐,趙康
(西安理工大學材料科學與工程學院,陜西西安710048)
摘要:圍繞污水處理領域的前沿技術,設計了碳包覆CeO2-Co3O4復合材料制備及其催化性能優化綜合實驗。實驗融合了溶劑熱反應、溶膠凝膠反應和模板法,制備出具有“核殼”及“搖鈴”結構的復合材料;借助XRD、TEM、SEM、FTIR、N2吸附-解吸附測試等表征手段剖析了材料的結構特性。研究了材料催化活化過一硫酸鹽(PMS)降解亞甲基藍(MB)的性能;建立了材料結構與性能間的聯系,并基于最優材料,提出了其催化機制。該實驗訓練了學生對化學及材料學科知識的理解和綜合運用能力,有助于培養他們的實踐能力、自主創新意識和科學思維能力。
關鍵詞:CeO2-Co3O4;碳;核殼及搖鈴結構;亞甲基藍;PMS活化;實驗設計
合成染料是全球水污染的主要來源之一。其中,亞甲基藍(MB)、亞甲基紅(MR)和羅丹明B(RhB)等合成染料,在紡織、化妝品、皮革、制藥、涂料以及造紙等眾多行業應用廣泛。這些染料即使在水中的含量極低(小于1ppm),也會使水體變得不宜飲用[1]。例如,MB不僅能引發高血壓、皮膚刺激以及腸胃不適等癥狀,更與癌癥和腎衰竭等嚴重疾病存在關聯。此外,由于這類合成染料具有較強的持久性和抗自然降解能力,它們在水環境中的存在還會對天然水體和生態平衡造成不可逆轉的破壞[2]。
近年來,基于過一硫酸鹽(PMS)的水處理技術發展迅速。PMS分解可產生具有高氧化活性的硫酸根自由基(SO–)等活性氧(ROS),因此被認為是降解水中有機污染物的有效手段[3]。有研究表明,Fe、Co、Ni、Mn、Cu等過渡金屬元素的氧化物可作為催化劑,大幅提高PMS的分解速率[4]。并且,得益于Ce3+/Ce4+的氧化還原特性和豐富的氧空位,將這些催化劑與CeO2復合,可增加吸附和反應位點、加速電子轉移,從而加快有機污染物的氧化降解[5-7]。例如,Liang等證實了Co3O4-CeO2復合材料在活化PMS降解三氯乙烯方面的優越性,并揭示了鈷、鈰離子間的協同效應[5]。但催化劑的尺寸一般都在納米級,在溶液中更容易團聚和腐蝕,嚴重限制了其性能發揮。人們發現,在金屬氧化物外部包覆碳材料,不僅可以阻止活性物質的聚集、減少金屬離子的溶解溢出,還能提供電子傳輸的快速通道,進而提高材料的氧化還原催化活性[8-9]。另一方面,催化材料的結構強烈影響著它的性能,中空及多孔結構有利于暴露更多活性位點、促進反應物富集,并減少SO–等ROS的擴散距離,提高反應效率。同時,反應發生在中空及多孔結構內部,還存在空間限域作用,能夠顯著改變化學反應過程,提高有機物的降解效率[10-13]。
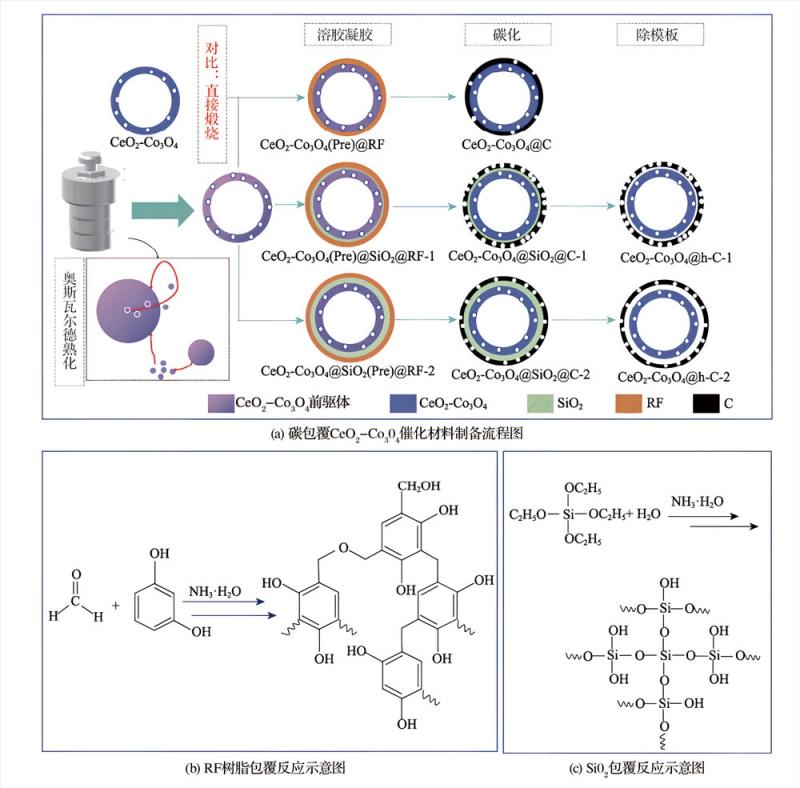
如圖1所示,在本實驗中,首先采用溶劑熱法制備中空Co3O4-CeO2前驅體,然后在其表面包覆間苯二酚-甲醛(RF)樹脂,再碳化轉化為“核殼”結構材料,其中Co3O4-CeO2作為核、碳作為殼。同時,通過引入不同厚度的二氧化硅(SiO2)中間層作為模板,經過碳化和去除模板,制備了具有不同“間隙”尺寸的“搖鈴”結構材料。最后,通過系統的測試、分析和合理對比,建立了材料結構與其性能之間的內在聯系,優化了其活化PMS降解MB的效能。本實驗涵蓋了納米材料的制備、表征與應用,不但能提高學生的實驗設計能力,還有利于培養學生的科學思維和創新意識。
1 實驗設計
1.1實驗原理
碳包覆CeO2-Co3O4催化材料的制備。如圖1(a)所示,以檸檬酸鹽為螯合劑和沉淀劑,與Ce3+/Co2+離子形成檸檬酸鹽沉淀物。在溶劑熱和堿性條件下,這些沉淀物水解并通過奧斯瓦爾德熟化過程形成具有中空結構的CeO2-Co3O4前驅體。接下來,以該前驅體為“核”,根據設計需要,通過溶膠-凝膠法在其表面包覆SiO2模板層和/或RF樹脂層,最終通過碳化及刻蝕獲得不同結構的碳包覆CeO2-Co3O4催化材料。RF樹脂層和SiO2層包覆反應示意圖見圖1(b)和(c)。
圖1碳包覆CeO2-Co3O4催化材料制備流程圖及RF樹脂和SiO2包覆反應示意圖
1.2 實驗試劑與儀器
主要試劑:七水合氯化鈰(CeCl3·7H2O)、六水合氯化鈷(CoCl2·6H2O)、二水合檸檬酸三鈉(C6H5O7Na3·6H2O)、尿素(CH2N2O)、乙二醇(C2H6O2)、氨水(NH3·H2O,28%)、正硅酸乙酯(C8H20O4Si)、3-氨基丙基三乙氧基硅烷(C9H23NO3Si)、間苯二酚(C6H6O2)、甲醛(CH2O,30%)、氫氧化鈉(NaOH)、亞甲基藍(MB,C16H18N3ClS·3H2O)、過一硫酸氫鉀復合鹽(PMS,2KHSO5·KHSO4·K2SO4)、叔丁醇(C4H10O)、對苯醌(C6H4O2)、L-組氨酸(C6H9N3O2)、乙醇(C2H6O)。
主要設備與儀器:分析天平、磁力攪拌器、超聲波清洗儀、高速離心機、反應釜、電熱恒溫鼓風干燥箱、管式電阻爐、X射線衍射儀(XRD,XRD-7000)、掃描電子顯微鏡(SEM,JEOL,JEM-6700F)、透射電子顯微鏡(TEM,JEOL,JEM-3010)、紅外光譜儀(FTIR,Nicolet,Avatar360)、比表面及孔徑分析儀(Micromeritics,ASAP2020)、電感耦合等離子體原子發射光譜儀(ICP,PerkinElmer,Optima8300)、紫外-可見光分光光度計(西安比朗,7601MC)等。
1.3 實驗步驟
1.3.1 材料制備
中空Co3O4-CeO2前驅體制備。將0.75mmolCeCl3·7H2O和0.75mmolCoCl2·6H2O溶解在5mL去離子水中,加入1.5mmol檸檬酸三鈉,配制成溶液A;將0.6g尿素溶解于20mL乙二醇中配制成溶液B。將溶液A與溶液B混合均勻后,轉移至50mL反應釜內,并在200℃的鼓風烘箱中保溫10h;反應釜自然冷卻后,離心收集沉淀物,用去離子水和無水乙醇分別清洗三次后放入60℃烘箱中干燥12h,獲得Co3O4-CeO2前驅體,簡記為:Co3O4-CeO2(Pre)。
“搖鈴”結構復合材料制備。將水熱制備的前驅體樣品分散到由128mL乙醇、32mL去離子水和3mL氨水組成的混合液中,分別加入0.75mL及1.5mLTEOS,并在室溫下攪拌6h;向體系加入0.15mLKH-550,繼續攪拌2h;最后,依次加入0.2g間苯二酚和0.28mL的甲醛,攪拌12h,獲得包覆產物(分別命名為:Co3O4-CeO2(Pre)@SiO2@RF-1和Co3O4-CeO2(Pre)@SiO2@RF-2)。離心收集沉淀物并干燥后,將其置于管式爐中,在N2氣氛下500℃碳化2h,升溫速率為5℃/min。將碳化產物浸泡于1.5mM的NaOH溶液中,于40℃條件下刻蝕4h,將產物收集、清洗和干燥。其中,TEOS添加量為0.75mL和1.5mL時,材料分別命名為:Co3O4-CeO2@h-C-1和Co3O4-CeO2@h-C-2。
“核殼”結構復合材料制備。將水熱制備的前驅體樣品分散到128mL乙醇、32mL去離子水和3mL氨水組成的混合液中。加入0.15mLKH-550并攪拌2h后,依次加入0.2g間苯二酚和0.28mL的甲醛,攪拌12h,獲得包覆產物(命名為:Co3O4-CeO2(Pre)@RF)。離心收集沉淀物并干燥后,將其置于管式爐中,在N2氣氛下500℃碳化2h,所制備材料命名為:Co3O4-CeO2@C。
中空Co3O4-CeO2制備。作為對比,本實驗直接將水熱制備的前驅體樣品置于管式爐中,在N2氣氛下500℃加熱2h,升溫速率為5℃/min,獲得樣品命名為:Co3O4-CeO2。
1.3.2催化性能測試
量取50mL的MB(50mg/L)溶液置于雙層石英管中,石英管夾層中通入循環水,使反應溫度始終保持在25℃。稱取15.0mg催化劑分散于MB溶液中,攪拌30min,以達到吸附平衡。向混合液中加入30.1mgPMS,隨后每隔一定時間取1.5mL反應混合液,立即用0.22μm的濾頭過濾。取1mL濾液加入到含甲醇猝滅劑的去離子水溶液中(5mL),然后使用紫外-可見光分光光度計記錄MB溶液在664nm波長處的吸光度,并通過吸光度的變化來反映MB溶液濃度的變化。MB的降解率η可通過以下公式計算
式中:A0為MB溶液的初始吸光度,At為t時刻MB溶液的吸光度。
1.3.3自由基清除實驗
在催化劑與MB溶液達到吸附平衡后,向體系中添加ROS清除劑,再加入PMS開始催化反應,通過MB降解率的變化來確定對降解反應過程起主要作用的ROS。實驗使用乙醇(EtOH)作為硫酸根自由基(SO4–)和羥基自由基(·OH)的清除劑,叔丁醇(TBA)作為羥基自由基(·OH)的清除劑,對苯醌(p-BQ)作為超氧自由基(O–)的清除劑,L-組氨酸(L-H)作為單線態氧(1O2)的清除劑。
2 實驗結果與討論
2.1XRD分析
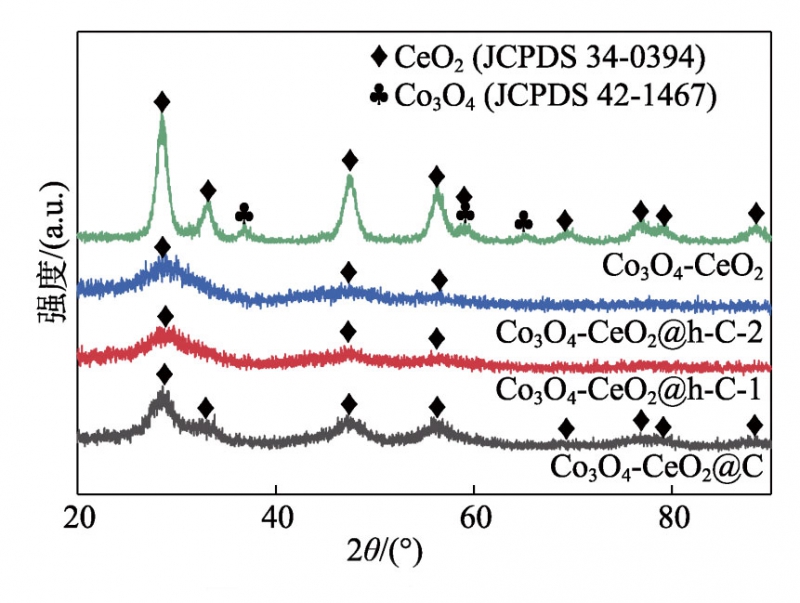
圖2為所制備材料的XRD譜圖,對于Co3O4-CeO2,在2θ=28.5°、33.1°、47.5°、56.2°、58.9°、69.4°、76.8°、
79.0°和88.4°處的衍射峰歸屬于立方相CeO2(JCPDSNO.34-0394)的(111)、(200)、(220)、(311)、(222)、(400)、(331)、(420)和(422)晶面;在2θ=36.8°、
59.1°和65.1°處的衍射峰歸屬于立方相Co3O4(JCPDSNO.42-1467)的(311)、(511)和(440)晶面。這說明前驅體直接在氮氣下煅燒,可成功轉化為Co3O4和CeO2的復合物。此外,通過ICP測試確定了Co3O4-CeO2中Co與Ce的摩爾比約為0.51∶1,所以譜圖中Co3O4的衍射峰較低。而在Co3O4-CeO2@C、Co3O4-CeO2@h-C-1和Co3O4-CeO2@h-C-2的XRD譜圖中,雖然能觀察到歸屬于CeO2的衍射峰,但這些峰的強度變低、峰寬變寬,這是因為前驅體表面的包覆層在煅燒過程中有效抑制了氧化物的晶粒生長,并且SiO2的抑制效果明顯強于RF樹脂[14]。同時,在核殼及搖鈴型材料的XRD譜圖中均觀察不到Co3O4的衍射峰,表明Co3O4高度分散于CeO2中。
圖2材料的XRD譜圖
2.2 TEM和SEM分析
圖3(a)的TEM照片顯示Co3O4-CeO2前驅體具有不規則的“生姜”狀輪廓,尺寸約為100~500nm,并且其內部呈現出明顯的空腔結構。接下來,采用溶膠凝膠法對Co3O4-CeO2前驅體進行表面包覆。當僅向反應體系中加入間苯二酚和甲醛時,從圖3(b)可以觀察到Co3O4-CeO2前驅體外圍形成了致密且連續的殼層,厚度約為15~20nm,說明RF樹脂層包覆成功。而當依次加入TEOS以及間苯二酚/甲醛后,在相應的TEM照片中(見圖3(c)和(d)),前驅體顆粒表面出現了襯度不同的雙殼層結構,由內到外分別為SiO2模板層(圖中箭頭標記處)和RF樹脂層。值得注意的是,隨TEOS添加量由0.75mL增加到1.5mL,SiO2模板層的厚度也由5~8nm提高到約24~30nm。
圖3材料的TEM照片
以上樣品經過N2條件下500℃處理及去除模板(對于搖鈴結構材料)后,其TEM照片見圖3(e)—(h)。首先,Co3O4-CeO2材料的整體形態和結構(見圖3(e))與前驅體保持一致。類似地,相比于碳化前,Co3O4-CeO2@C依然保持著良好的核殼結構(見圖3(f))。而 與之形成鮮明對比的是,在Co3O4-CeO2@h-C-1和Co3O4-CeO2@h-C-2的TEM照片(見圖3(g)和(h))中,Co3O4-CeO2內核與C外殼間產生了明顯的間隙,但Co3O4-CeO2@h-C-2的間隙更大,這得益于其制備過程中采用了更厚的SiO2模板層[15]。
圖4展示了所制備核殼及搖鈴結構材料的SEM照片,可以觀察到所有樣品都由顆粒聚集而成,但對于Co3O4-CeO2@h-C-1和Co3O4-CeO2@h-C-2,產物中有少量顆粒發生了破碎,這是由于部分碳殼層較薄,在SiO2模板去除后無法自支撐造成的。但同時,該結果也證明其內部是中空結構。以上結果充分證明,本實驗通過簡單控制制備過程中TEOS的用量(0、0.75、1.5mL),可成功制備具有“核殼”及“搖鈴”結構的碳包覆Co3O4-CeO2復合材料,并能有效調控后者的間隙尺寸,因此具有很高的便利性和靈活性。
2.3 FTIR分析
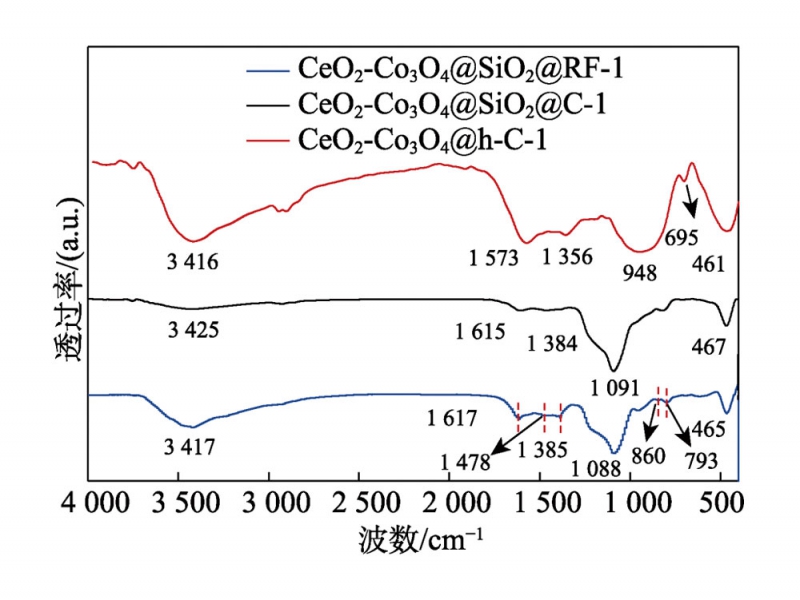
本實驗以Co3O4-CeO2@h-C-1為例,利用FTIR光譜對制備過程中各步驟產物的表面官能團進行了分析(見圖5)。在包覆產物CeO2-Co3O4(Pre)@SiO2@RF-1的譜圖中,3417cm–1處的寬峰歸屬于-OH的拉伸振動峰,1617cm–1、1478cm–1和1385cm–1處的譜峰來自于RF樹脂中苯環的骨架振動,793cm–1和860cm–1處的峰對應于苯環上的鄰、對位取代[16]。而1088cm–1處出現的吸收峰歸因于Si—O—Si的對稱拉伸振動,465cm–1處的吸收峰對應于Ce—O鍵的拉伸振動。以上結果證明了RF樹脂和SiO2的成功包覆。碳化后,CeO2-Co3O4@SiO2@C-1樣品的FTIR譜圖中除了1091cm–1和467cm–1處的Si—O—Si鍵和Ce—O鍵的拉伸振動外,其他峰的強度大大削弱,表明碳化過程中樣品表面的大量羥基及有機基團發生分解。對于Co3O4-CeO2@h-C-1,譜圖中歸屬于Si—O—Si鍵的吸收峰消失,表明SiO2已被溶解去除。而948cm–1處出現的特征峰可能是在NaOH刻蝕過程中形成了一定量的Co—O—Si鍵,這有利于樣品表面缺陷的形成。此外,695cm–1處的特征峰則歸因于Co—O鍵,在模板去除后顯現了出來[5,17]。
圖4材料的SEM照片
圖5樣品的FTIR譜圖
2.4 比表面積分析
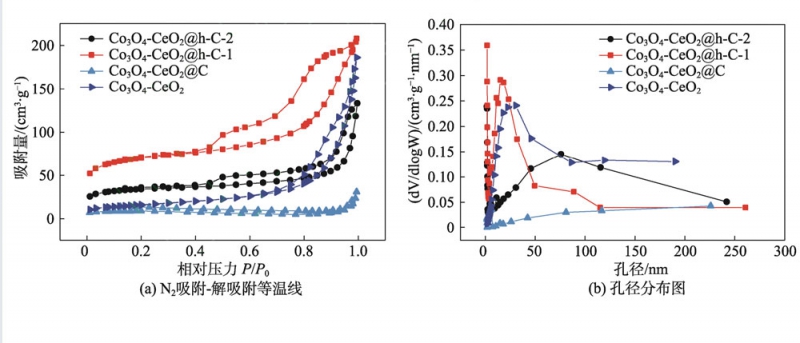
圖6(a)為樣品的N2吸附-解吸附等溫線,圖中所有曲線均可歸為第Ⅳ類等溫線,并出現了明顯的滯后環,說明樣品中存在大量介孔結構[5]。測試結果顯示,CeO2-Co3O4的比表面積約為55.8m2/g,而CeO2-Co3O4@C的比表面積僅為24.8m2/g,這是因為碳殼層對內核材 料的緊密包覆阻礙了外界N2的進入。相反,對于具有“搖鈴”結構的Co3O4-CeO2@h-C-1和Co3O4-CeO2@h-C-2,其比 表面積分別增加到222.1和107.1m2/g,這是因為煅燒 過程中SiO2對Co3O4和CeO2燒結及晶粒長大起到抑 制作用,降低了內核材料的比表面積損失[14]。另一方 面,在溶膠凝膠過程中,TEOS水解/縮合產生的正硅酸低聚物并未完全沉積到SiO2模板層中,而是部分進 入了后續包覆的RF樹脂層中,經過碳化和刻蝕后,在碳殼層中留下了大量孔洞,不但提供了更多的比表面積,還有利于材料的內外聯通[18]。值得注意的是, Co3O4-CeO2@h-C-1的比表面積遠高于Co3O4-CeO2@ h-C-2,這可能是因為后者在制備過程中加入的TEOS更多,導致正硅酸縮合和沉積反應更完全,從而使造孔效果下降。圖6(b)顯示了材料的孔徑分布圖,證明各材料均富含大量孔道,且分布較寬。
圖6材料的N2吸附-解吸附等溫線及孔徑分布圖
2.5 催化性能及構效關系分析
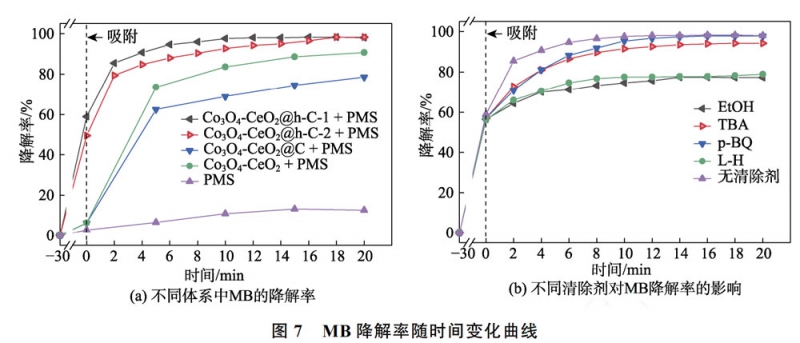
實驗以MB為模擬污染物,考察不同結構材料活化PMS的催化性能。首先,如圖7(a)所示,PMS自身降解MB的效率僅為12.6%。而將所制備材料與MB溶液攪拌30min達到吸附平衡后,Co3O4-CeO2@h-C-1、Co3O4-CeO2@h-C-2、CeO2-Co3O4@C和CeO2-Co3O4對MB的吸附率分別為58.9%、48.5%、6.2%和6.3%。其中,“搖鈴”型結構材料的吸附率較高,這是因為其較高的比表面積。此外,雖然CeO2-Co3O4@C的比表面積小于CeO2-Co3O4,但其對MB的吸附率與后者相當,這得益于CeO2-Co3O4@C中碳殼層與MB分子間的疏水-疏水相互作用。當加入PMS后,Co3O4-CeO2@h-C-1和Co3O4-CeO2@h-C-2體系中的MB迅速降解,反應2min后,降解率分別達到85.4%和79.4%;繼續延長反應至20min,MB幾乎被完全降解。而對于CeO2-Co3O4@C,其在20min時可降解78.5%的MB,低于CeO2-Co3O4的90.7%。以上結果表明,Co3O4-CeO2@h-C-1的催化活性最優,這是因為其具有最高的比表面積,有利于內部活性位點的暴露和物質的富集與交換,并且較小的“間隙”尺寸有利于形成更顯著的空間限域作用。同時,材料晶粒尺寸的降低容易產生更多的表面氧空位(Ov)等缺陷,有利于PMS的活化[14,19]。相反,CeO2-Co3O4@C具有最差的催化性能,這歸因于其致密的碳包覆層,阻礙了材料的內外物質交換。由此可見,在材料組成基本不變的情況下,可以通過材料結構的調控優化其催化性能。
2.6 活性氧與催化機理分析
選取催化性能最優的Co3O4-CeO2@h-C-1為研究對象,利用清除劑來確定MB降解反應中的主要ROS。如圖7(b)所示,當體系中加入TBA和p-BQ時,反應前期MB的降解速率有所降低,但反應延長到20min
后,MB的降解率下降并不明顯。說明·OH和O–在體
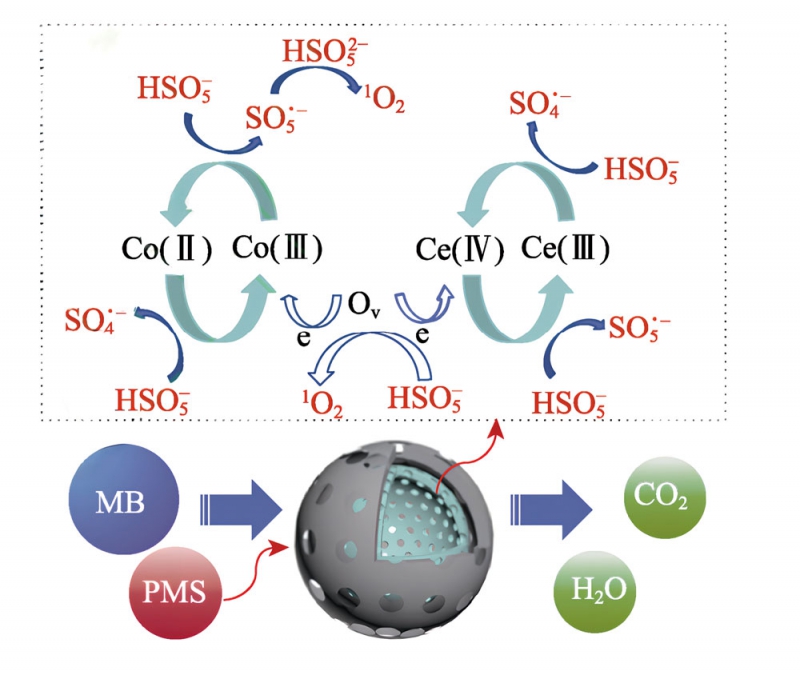
系中的作用很小。而加入EtOH和L-H后,MB降解率分別從98.1%降低到77.3%和79.0%。考慮到Co3O4-CeO2@h-C-1對MB的吸附率較高(58.9%),可以判定SO4–和1O2是體系中起主要作用的ROS[8,13]。根據上述結果,提出了Co3O4-CeO2@h-C-1體系降解MB的催化機理,如圖8所示。催化劑表面的Co(Ⅱ)/Ce(Ⅲ)可與PMS反應生成SO4–(式(2)),Co(Ⅲ)/Ce(Ⅳ)同時也可以被還原再生為Co(Ⅱ)/Ce(Ⅲ)(式(3)),完成催化循環[3-4]。催化劑中的Ov等缺陷有利于促進PMS分子吸附,Ov的富電子特性也可促進Co(Ⅲ)/Ce(Ⅳ)與Co(Ⅱ)/Ce(Ⅲ)之間的氧化還原循環。
此過程中,所形成的SO—會進一步與H2O反應,生
成少量·OH(式(4))和O–(式(5)、(6))。另一方面,
PMS通過自身分解,或者奪取Ov的電子均可生成1O2(式(7)—(9))。最終,反應生成的ROS(主要為1O2和SO4–)將MB分子氧化降解[19-20]。
圖8Co3O4-CeO2@h-C-1催化活化PMS降解MB的反應機理圖
3 結語
本綜合實驗融合了溶劑熱法、溶膠凝膠法和模板法,成功制備出具有“核殼”與“搖鈴”結構的碳包覆Co3O4-CeO2復合材料。通過TEM、SEM、FTIR、N2吸附-解吸附實驗等測試手段對材料進行了表征。催化實驗結果表明,具有“搖鈴”型結構的材料的催化性能明顯優于“核殼”型材料。其中,“間隙”較小且比表面積最大的Co3O4-CeO2@h-C-1對MB的吸附率高達58.9%,并在反應2min后,可去除85.4%的MB。通過對實驗結果的綜合分析,可以引導學生理解材料結構與性能之間的聯系。該實驗跨越了化學、材料科學、環境科學等多個學科領域,不僅使學生掌握了先進的綜合實驗技能,還有助于培養他們的自主創新意識和科學思維能力,為學生將來投身科研工作奠定基礎。
參考文獻
[1]AHMED H R, KAYANI K F. A comparative review of Fentonlike processes and advanced oxidation processes for Methylene Blue degradation[J]. Inorganic Chemistry Communications, 2024, 170(P3): 113467.
[2] MODI S, YADAV V K, GACEM A, et al. Recent and emerging trends in remediation of methylene blue dye from wastewater by using zinc oxide nanoparticles[J]. Water, 2022, 14(11): 1749.
[3]代朝猛 ,劉仟,段艷平,等 . 活化過—硫酸鹽技術降解環境有機污染 物的研究進展 [J]. 環境科學研究,2022,35(1): 141–149.
[4]FARSHID G, MAHSA M. Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants: Review[J]. Chemical Engineering Journal, 2016(310): 41–62.
[5]LIANG X T, LI Y X, CUI L Y, et al. Morphology control of Co3O4/CeO2 heterojunctions toward efffcient peroxymonosulfate activation for trichloroethylene removal: Effect of oxygen precursors[J]. Journal of Water Process Engineering, 2024(59): 105016.
[6] LI Z D, LIU D F, ZHAO Y X, et al. Singlet oxygen dominated peroxymonosulfate activation by CuO-CeO2 for organic pollutants degradation: Performance and mechanism[J]. Chemosphere, 2019(233): 549–558.
[7]LI J, GOU G, ZHAO H L, et al. Efficient peroxymonosulfate activation by CoFe2O4-CeO2 composite: Performance and catalytic mechanism[J]. Chemical Engineering Journal, 2022, 435(P1): 134840.
[8]LIU Z C, ZHONG Y, CHEN L, et al. Co3O4/CuO@C catalyst based on cobalt-doped HKUST-1 as an efffcient peroxymonosulfate activator for pendimethalin degradation: Catalysis and mechanism[J]. Journal of Hazardous Materials, 2024(478): 135437.
[9]NIU J L, HE T F, CHENG J H. In-situ synthesis of carbon-coated Co3O4 nanowire for efffcient activation of peroxymonosulfate while reducing ion leaching through protection and secondary enrichment[J]. Chemical Engineering Journal, 2024(500): 157301.
[10]LI B W, ZENG H C. Architecture and preparation of hollow catalytic devices[J]. Advanced Materials, 2019, 31(38): 1801104.
[11] SHI B Y, YU H T, GAO S G, et al. Copper complex supported on hollow porous nanosphere frameworks with improved catalytic activity for epoxidation of oleffns[J]. Microporous and Mesoporous Materials, 2020(294): 109890.
[12]劉芳,陳淑華, 李晨星,等 .基于非金屬中空球臭氧催化氧化協同光催化系統處理新污染物實驗設計[J]. 實驗技術與管理, 2022, 39(12): 10–17.
[13]錢坤,林希典,姜晶.空間限域策略強化高級氧化技術去除 水 中有機污染物研究進展[J/OL]. 中國環境科學, 1–6. (2024-12-17) [2025-01-16].https://doi.org/10.19674/j.cnki.issn1000-6923.20241216.006.
[14]MENG Q N, LIANG X L, GUO H, et al. Facile preparation of CeO2 and Co3O4 hollow composite with abundant oxygen vacancies and high surface area for improved acetone sensing[J]. Materials Research Bulletin, 2024(170): 112551.
[15]CUI Z M, CHEN Z, CAO C Y, et al. A yolk–shell structured Fe2O3@mesoporous SiO2 nanoreactor for enhanced activity as a Fenton catalyst in total oxidation of dyes[J]. Chemical Communication, 2013(49): 2332–2334.
[16] MENG Q N, WANG K, TANG Y F, et al. One-pot synthesis of Fe2O3 loaded SiO2 hollow particles as effective visible light photo-Fenton catalyst[J]. Journal of Alloys and Compounds: An Interdisciplinary Journal of Materials Science and Solid-state Chemistry and Physics, 2017(722): 8–16.
[17]HAO S M, YU M Y, ZHANG Y J, et al. Hierarchical mesoporous cobalt silicate architectures as highperformance sulfate-radical- based advanced oxidization catalysts[J]. Journal of Colloid and Interface Science, 2019(545): 128–137.
[18]MO Y H, DUJ, LYU H J, et al. N-doped mesoporous carbon nanosheets for supercapacitors with high performance[J]. Diamond & Related Materials, 2021(111): 108206.
[19] WU L Y, YU Y B, ZHANG Q, et al. A novel magnetic heterogeneous catalyst oxygen-defective CoFe2O4-x for activating peroxymonosulfate[J]. Applied Surface Science, 2019(480): 717–726.
[20]劉芳,劉嘉梁,安蓓雅,等.自然光條件下光自芬頓/PMS 協同體系處理新污染物的實驗設計[J]. 實驗技術與管理 , 2024, 41(1): 26–36.








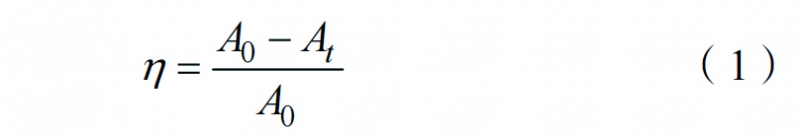

















 玻纖含量對長玻纖...
玻纖含量對長玻纖... 鈣鈦礦薄膜的均勻...
鈣鈦礦薄膜的均勻... 用于光伏板靜電除...
用于光伏板靜電除... 聚砜醫療干粉吸入...
聚砜醫療干粉吸入...





